
Quantum computing is steadily advancing from theory to application. One of the most promising near-term use cases is in quantum chemistry – where quantum computers have the potential to outperform classical computers in simulating molecules and materials. But even with today’s best quantum devices, making real chemical insights has been a challenge – until now.
In this post, we demonstrate how the Kvantify Chemistry Quantum Development Kit (QDK) enables accurate and affordable quantum chemistry calculations on IQM’s 16-qubit Sirius (star topology) and 20-qubit Garnet (crystal square lattice topology) superconducting quantum processors, accessed via IQM Resonance, the quantum cloud platform.
By replicating and extending a 2023 IBM Quantum study on the dissociation of butyronitrile, Kvantify’s QDK showcases how real-world quantum hardware can now be harnessed for scalable, high-fidelity chemical simulations.
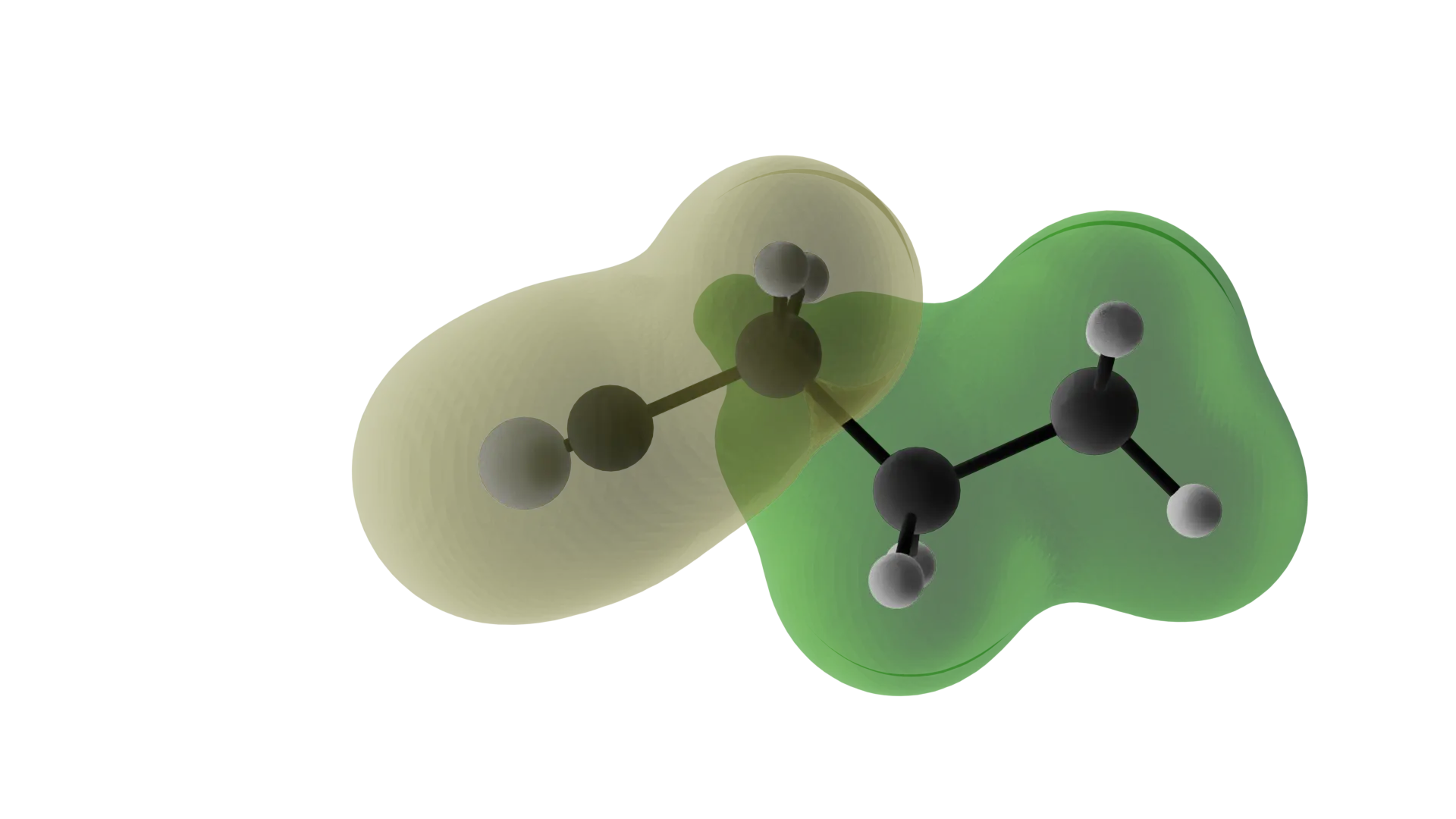
Image Credit: Kvantify
Visualization of electron densities and chemical bonds during the nitrogen dissociation process:The moving atom is the nitrogen atom, carbon atoms are shown in dark gray, and hydrogen atoms in white. (Lewis) electron pairs are indicated as black connections between the atoms, and under the bond-breaking process, the (triple) bond between nitrogen and carbon is broken. A more realistic picture of the chemical bonds is shown by the light-green and dark-green electron densities derived from quantum-chemical calculations. The light-green part of the electron density is the part for which an effective Hamiltonian is constructed for the calculations on quantum hardware, and the dark-green part is the (static) environment that enters into the effective Hamiltonian.
The simulation of molecular systems such as butyronitrile traditionally requires extensive classical computation. However, as quantum processors surpass the 16–20 qubit range, classical simulation becomes a bottleneck due to the exponential scaling of quantum states. This is especially true in adaptive algorithms like ADAPT-VQE, which have been limited by their reliance on noise-free simulators for most of the computational workload.
Kvantify’s Chemistry QDK changes that. Built with scalability in mind, it allows quantum hardware to be used meaningfully in every step of a chemical calculation – not just for final energy evaluations. Kvantify has a patented FAST-VQE algorithm that runs adaptive operator selection on real quantum hardware, while a high-performance state vector simulator handles the optimisation step. This hybrid workflow dramatically reduces the cost and complexity of quantum chemistry calculations.
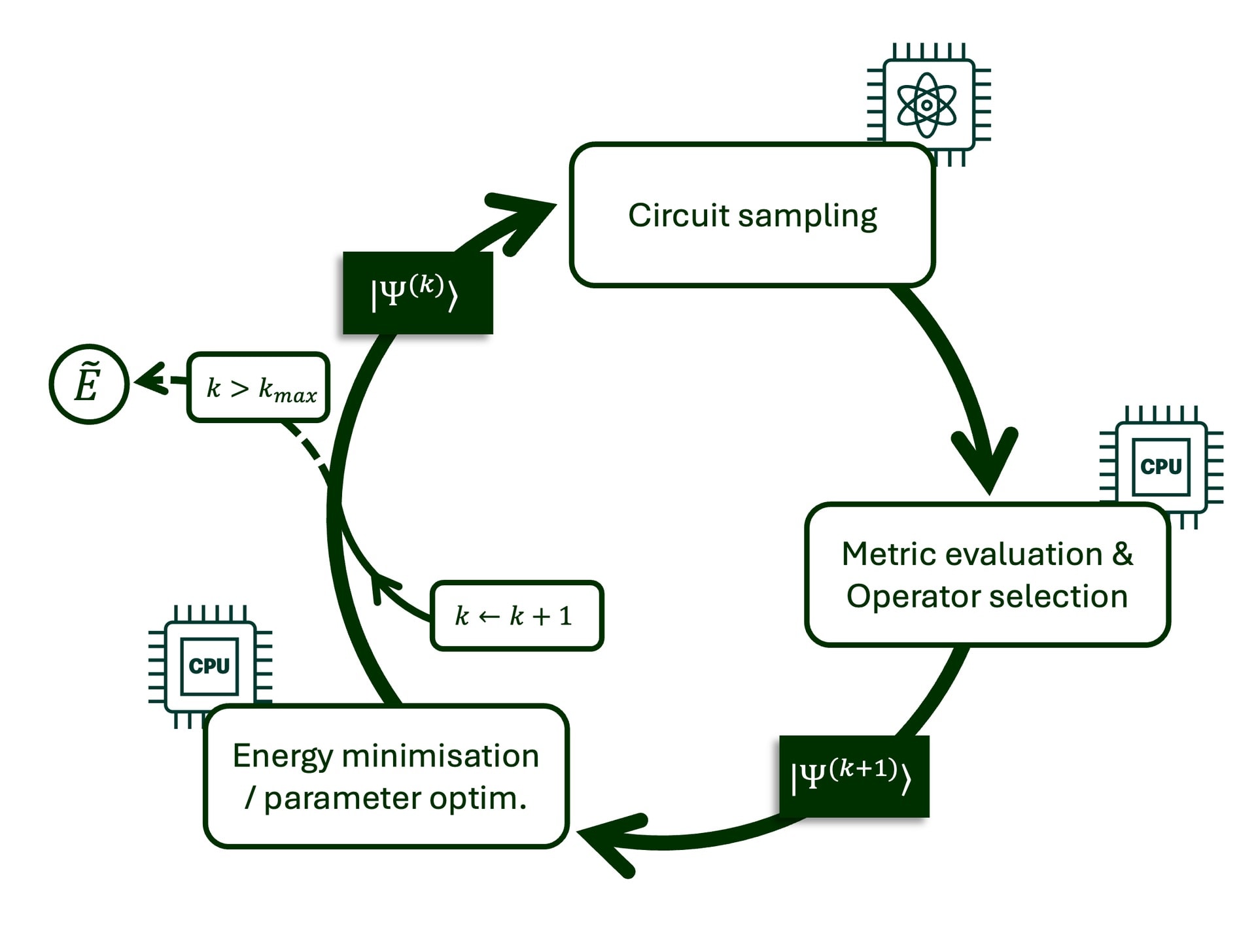
Image Credit: Kvantify
Visualization of the iteration process of Kvantify’s patented FAST-VQE method.
To test the QDK’s capabilities, Kvantify revisited IBM Quantum’s 2023 study of butyronitrile, a molecule with emerging importance in battery and solar cell research. IBM simulated the dissociation of the nitrile group using an 8-qubit minimal basis set and ADAPT-VQE. Kvantify reproduced and extended this work using IQM’s systems Sirius and Garnet, scaling up to 20 qubits and a realistic PCSEG-2 basis set.
The dissociation process involves gradually stretching the triple bond between the carbon and nitrogen atoms. At each step, the system’s total energy must be calculated, a process that previously required intensive classical simulations. With Kvantify’s QDK, the full energy profile (potential energy surface) was computed directly on IQM’s hardware, using 60 adaptive iterations per geometry point, corresponding to 2–3 seconds of quantum runtime per iteration.
Below are the results of simulating the dissociation of butyronitrile using IQM’s quantum systems and the Kvantify Chemistry QDK. The dissociation curves, showing the system’s energy as a function of the nitrogen–carbon bond distance, were computed across an increasing number of spin orbitals, corresponding to a growing number of qubits utilised.
In addition to the benefits of accessing larger active spaces through higher qubit counts, the simulations incorporate several improvements over the original 2023 study, including:
These enhancements contribute to more accurate and chemically relevant results and showcase the power of combining advanced quantum hardware with tailored quantum chemistry algorithms.
The plots below illustrate the computed potential energy surfaces (PES), i.e., the total energy of the chemical system at varying nitrogen–carbon bond distances, generated using the FAST algorithm on IQM’s systems Sirius and Garnet, as well as Kvantify’s exact, chemistry-specific state vector simulator. For validation, all results are compared to the exact CASCI reference calculated in the same orbital space, with the differences shown in the lower panels of the figures.
The simulations were performed with an increasing number of spin orbitals, effectively scaling up the problem size and eventually exhausting the capacity of each quantum processor. Across all configurations, we observe that the error trends are consistent between hardware and simulator.
As expected, the largest deviations occur near the dissociation limit, where the Hartree-Fock reference state deviates most from the true ground state and more adaptive iterations are required for convergence.
With 16 spin orbitals, fully utilising the Sirius system, the convergence is not as tight anymore with the same number of iterations, but convergence is still smooth towards the CASCI results. At 20 spin orbitals, which pushes the limits of the Garnet system, this effect becomes stronger. To improve accuracy at these larger system sizes, additional adaptive iterations would be necessary.
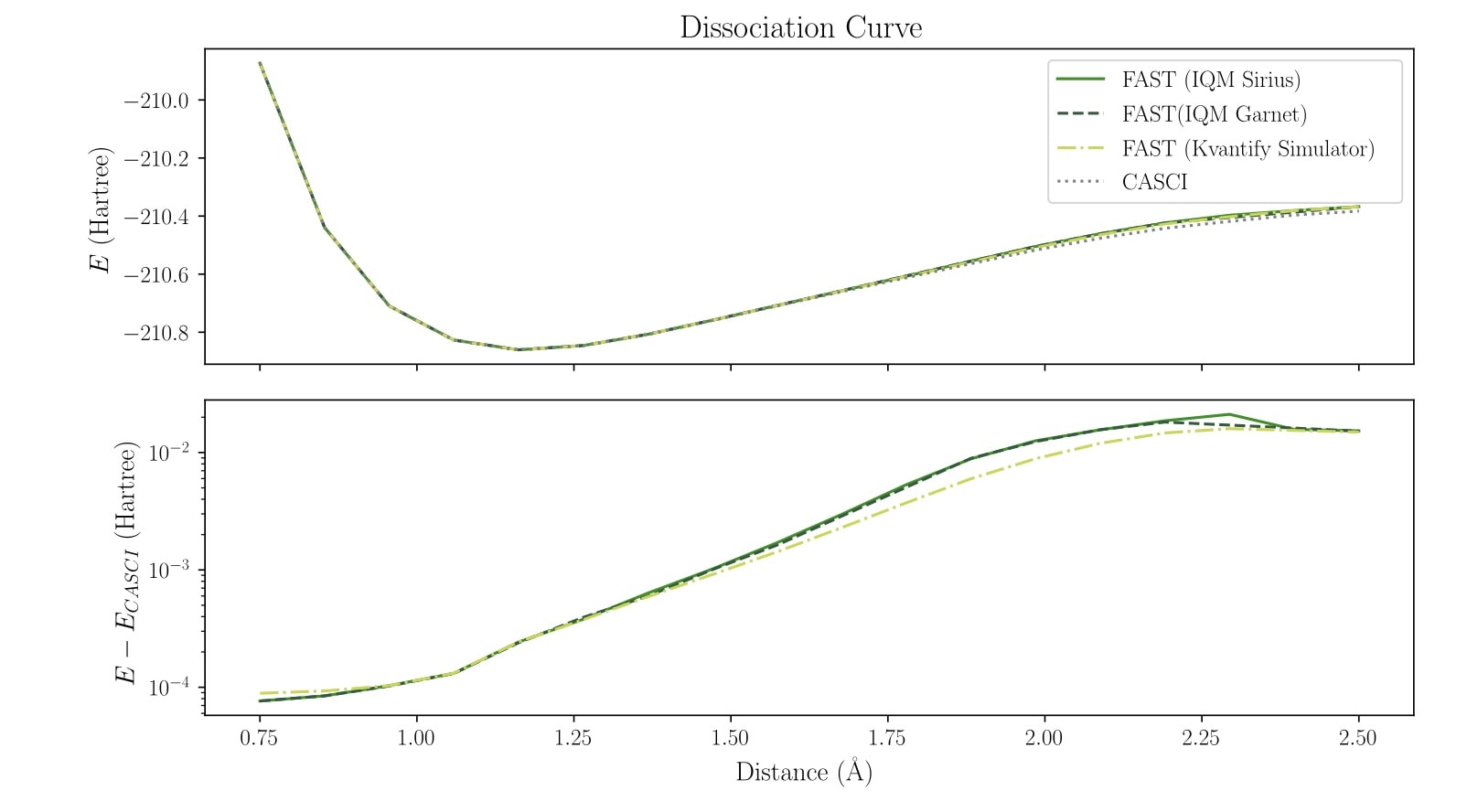
Dissociation curve computed with 12 spin orbitals on IQM Garnet and Sirius hardware
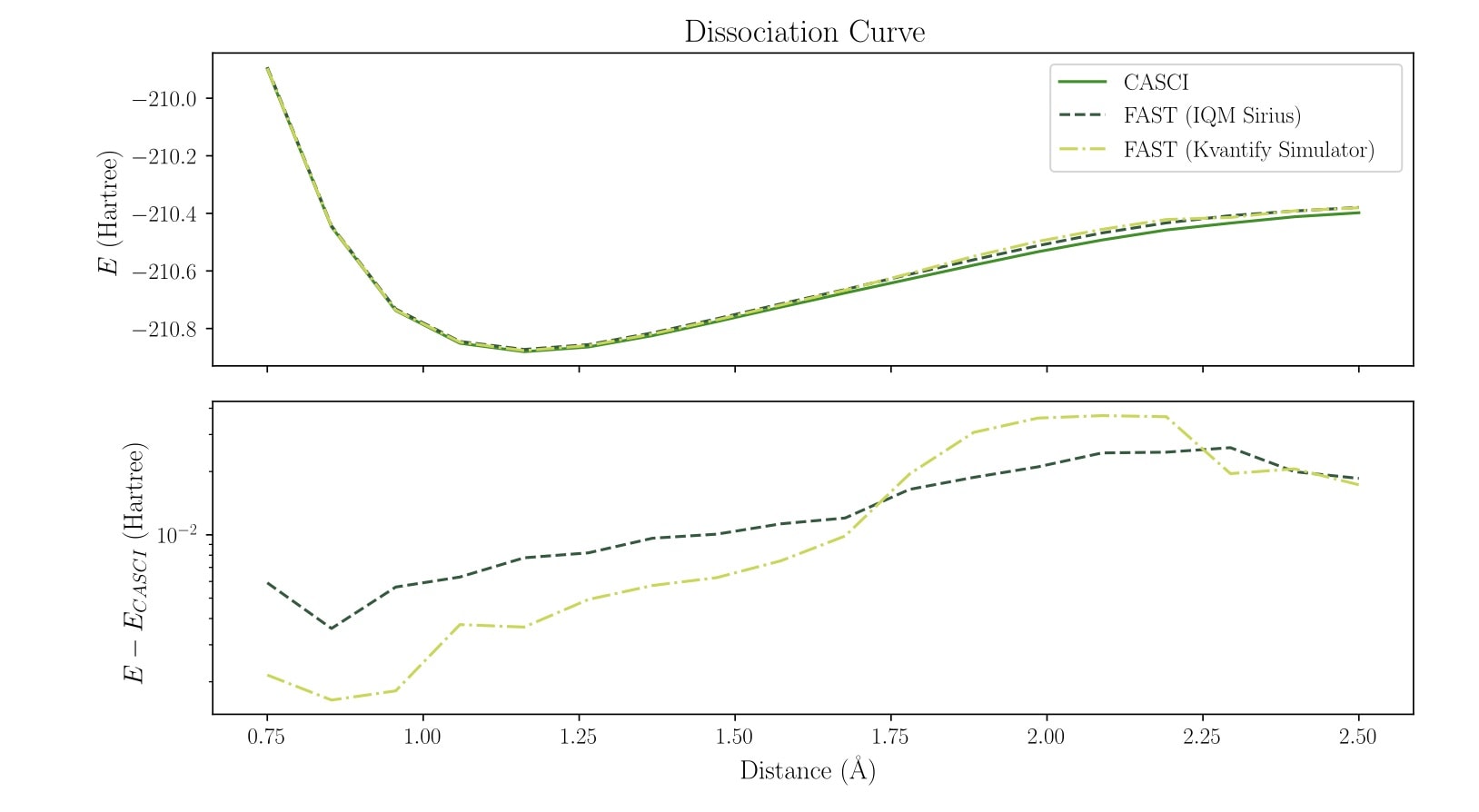
Dissociation curves computed with 16 spin orbitals on IQM Sirius hardware
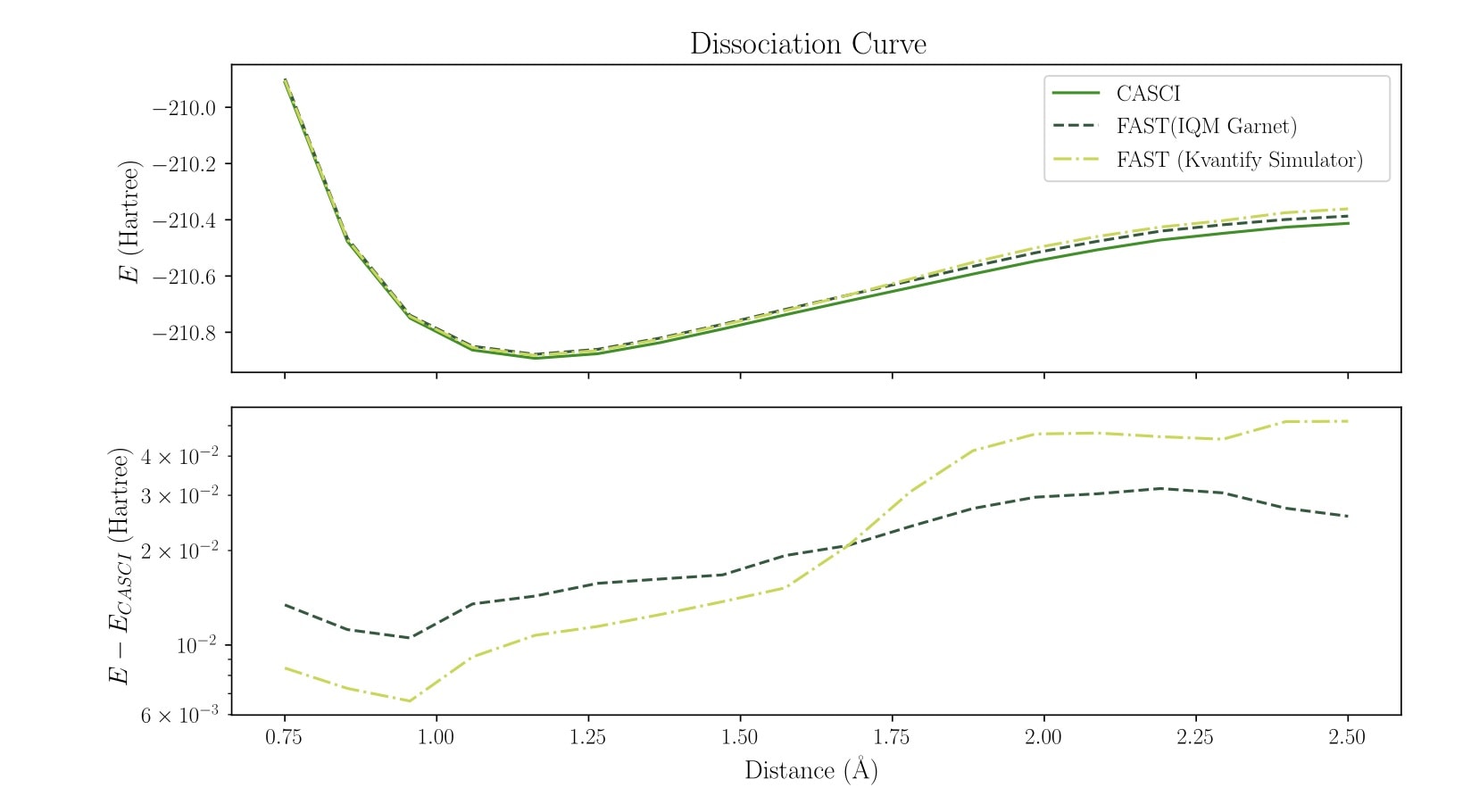
Dissociation curves computed with 20 spin orbitals on IQM Garnet hardware
Butyronitrile isn’t just a benchmark molecule – it’s an active research target. It’s being explored as a next-generation electrolyte for lithium-ion batteries and dye-sensitized solar cells (DSSCs). These technologies are vital for a sustainable energy transition but face major challenges in performance and stability across temperature extremes.
Butyronitrile’s favorable properties – low viscosity at low temperatures, and chemical stability at high voltages – make it a strong candidate for improved electrolyte formulations. Better understanding its behavior at the quantum level is key to unlocking future energy materials.
With Kvantify’s Chemistry QDK and IQM’s Resonance, researchers and industry professionals can now carry out scalable, affordable, and realistic quantum chemistry calculations on actual quantum hardware. The QDK simplifies the complexity of quantum algorithm design and lets chemists focus on insights – not infrastructure.
You want to test IQM’s quantum systems on via cloud. Get in touch here.
The Kvantify Chemistry QDK will soon be available for wider use. Visit Kvantify’s website to get in touch.
